病因
病因
病因:下丘脑分泌GHRH和SS,促进及调整垂体分泌GH,再进一步促进合成IGF-1和IGFBP-3共同作用于靶器官,促进生长和代谢,称此轴为生长轴。下丘脑又接受高级中枢神经传入的信息而受其影响。生长轴中任何环节有障碍均可引起生长迟缓导致身材矮小。
1.生长轴功能障碍的病因分类
(1)下丘脑-垂体先天异常:由于中枢神经系统的发育异常引起下丘脑-垂体的发育异常导致
生长激素缺乏。如全前脑缺乏或无脑,脑裂,视中隔发育不良,视神经发育不良。面部的畸形如单门齿脑中线发育不良,视神经伴透明隔发育不良,
唇裂腭裂等先天发育不良的部分患儿,伴有下丘脑缺陷和(或)垂体的GH或多种垂体激素分泌缺乏。单纯垂体发育不良不伴有脑发育障碍,曾报告有同胞兄弟和表亲同患病,为常染色体隐性遗传。空蝶鞍,为蝶鞍隔缺乏引起鞍上蛛网膜腔疝入鞍膜,使蝶鞍变形,垂体变平。中枢神经系统的一切先天病变凡影响下丘脑和垂体组织时,绝大部分患儿可产生下丘脑-垂体-IGF-1生长轴功能障碍导致身矮,或下丘脑-垂体多种激素的分泌障碍。
(2)破坏性病变:颅底骨折或出血,其他损伤包括出生时的缺血缺氧性脑病。
颅内肿瘤特别是颅咽管瘤,神经胶质瘤;
脑膜炎,颅内结核,
弓形体病,肉芽肿病,颅内
血管瘤等。对颅脑,眼及中耳部放射治疗,如中枢神经系统恶性肿瘤及白血病治疗时的头颅放疗,可影响生长轴的激素分泌。放射治疗开始的年龄,单次量,总剂量和每次放疗间隔的时间等,对下丘脑-垂体的影响不等。年龄小的较年龄大的危害大,放射量达到下丘脑-垂体的总剂量>1800~2000cGy时,发生GH轴障碍的发病率较高和开始时间较早。剂量<1800cGy可改变青春期GH
自发分泌的增高。剂量>2400cGy时GH
自发分泌减少,而刺激后仍可正常反应;剂量>2700cGy时
自发分泌和刺激后均受影响。如在短时间内大剂量的放射治疗则发生GH轴缺乏的危险更大。一般在放射治疗时经常联合化疗,化疗对颅内或脊髓内注射也是导致生长障碍的部分原因。
(3)特发性下丘脑-垂体功能减低(idiopathic
growth hormone deficiency,IGHD):多数病人下丘脑-垂体功能减低未能发现明显的病变,此类的问题多在下丘脑。常是散发的,有些为出生臀位产和出生时窒息,或产钳助产等,造成出生后缺血缺氧有关。
(4)遗传性下丘脑-垂体-生长轴功能障碍:遗传性身矮可有多种原因,McKusick曾分类为Ⅰ型为常染色体隐性遗传,Ⅱ型常染色体显性遗传,Ⅲ型性连锁遗传。以前多归属于特发性
生长激素缺乏(IGHD)。
产生GHRH-GH-IGF-1轴基因缺陷有GH1(GH-N)基因缺陷,该基因是产生hGH的基因,可有基因完全缺失,部分缺失或大小不同的片段缺失,甚至1~2bp的缺失。GH1完全缺失为IGHD1A型。国内曾报告一对姐妹GH缺乏,证实为GH1基因完全缺乏。常染色体隐性遗传为IGHD1B型,为GH1基因严重缺陷和点突变的杂合子,临床也是GH完全缺乏。常染色体显性遗传为GH1基因点突变,第3内含子一个碱基的突变可导致GHmRNA剪切掉第3外显子,使合成的GH缺少32~71位的氨基酸,缺少一个半胱氨酸不能形成分子内的二硫键,影响GH从分泌颗粒释放。X连锁IGHDⅢ型为家族性GHD伴有免疫球蛋白缺乏,可能涉及数个相连的基因缺失。
遗传性多种垂体激素缺乏多为常染色体隐性遗传或性连锁遗传,有GH、TSH、ACTH、LH和FSH缺乏,而PRL多正常或升高。若给予各种激素的释放因子试验垂体常能有反应,说明病变是在下丘脑。有些病儿家系证明为转录因子Pit-1基因的缺陷。Pit-1基因是GH,PRL和β-TSH基因的转录因子,此基因突变引起GH,PRL和TSH减少,有GHD同时有甲状腺功能减低。还发现有GHRH受体基因缺陷引起的GHD。
生长激素受体基因缺陷称GH不敏感症(growth hormone insensitivity,GHI):由于GH受体基因的缺失或突变使受体结构异常,GH不能与之结合,因而不能产生IGF-1,GH不能发挥作用,故称为GH不敏感。Laron综合征是首先发现的一种是由于先天性GH受体缺乏,临床常有
低血糖发生,生长障碍与GHD相似,血中GH浓度增高,而IGF-1非常低。经试验研究有发现血中GH结合蛋白(GH banding protein,GHBP)与核素标记GH结合能力下降。GHBP为GH受体的细胞外部分,说明GH受体有缺陷,对外源性GH无反应,不能促进生长。多数Laron综合征儿童的父母身高多在正常范围。本症可用IGF-1治疗。还有GH受体基因点突变,多发生在受体结构的胞外区。曾发现有的虽能产生受体但不能形成二聚体。此外受体后信息传递的异常。继发性GHD可因GH抗体或GH受体抗体的产生。营养不良或肝脏病虽血中GH正常而IGF-1产生减少亦产生GHD。所有生长激素不敏感症,血中GH的基础值均正常或高于正常。血中IGF-1,IGF-Ⅱ和IGFBP-3的浓度均减低。此外,有人将所有因GH缺乏和GH不敏感均归之为IGF-1缺乏类。
(5)精神性生长障碍:曾称为精神剥夺性侏儒。由于环境因素通过中枢神经系统产生抑郁情绪等,影响下丘脑-垂体生长激素的分泌减低,导致生长减慢。若能改变环境,心情舒畅,GH的分泌可以恢复正常,生长亦随之改善。
2.病因分类 根据下丘脑-GH-IGF轴功能缺陷,可分为:
(1)原发性:
①遗传:GH或GHRH基因异常或受体异常。
②特发性:下丘脑功能异常,神经递质-神经激素功能途径的缺陷。
③发育异常:垂体不发育、发育不良、空蝶鞍、视中隔发育不全等。
(2)继发性:
①肿瘤:颅咽管瘤、神经纤维瘤、错构瘤等。
②放射损伤:放疗后。
③头部创伤:产伤、手术损伤、颅底骨折等。
(3)IGF1缺陷:IGF1合成缺陷、IGF1受体缺陷等。
发病机制
发病机制:
1.生长激素和下丘-GH-IGF轴
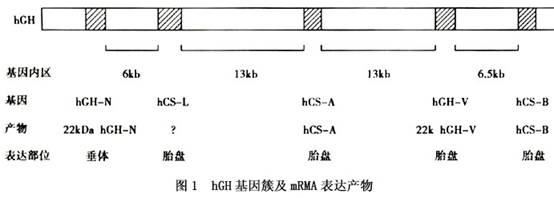
(1)生长激素(GH)的基因:GH是由垂体前叶嗜酸性粒细胞分泌的,含191个氨基酸,属非糖基化蛋白质激素,GH的半衰期为15~30min。人类GH基因簇由5个成员组成,定位于第17号染色体长臂q22~24区带。5个基因的排列顺序从5’至3’依次为hGH-N-hCS-L-hCS-A-hGH-V-hCS-B-3’。5个基因DNA序列有高度同源性,每个基因均含有5个外显子和4个内含子。其中hGH-N基因在垂体前叶嗜酸性粒细胞中表达,分泌生长激素。其他4个基因皆在胎盘滋养细胞中表达,与胎盘发育、胎儿生长有关(图1)。
(2)GH分泌和调节:在胎龄3个月内,垂体尚无GH分泌,其后血中GH水平逐步增高;至12周时,GH血浓度可达到60µg/L,30周时达130µg/L,以后GH浓度逐渐下降,出生时为30µg/L,以后进一步下降。GH分泌一般呈脉冲式释放,昼夜波动大,在分泌低峰时,常难以测到,血浓度常<5µg/L。深睡1h左右其GH分泌最为旺盛,在以后睡眠中,可见到较低峰。24h正常高峰节律为6~8次。
(3)GH的生理作用:GH的生理作用非常广泛,既促进生长,也调节代谢。其主要作用是:①促进骨生长;②促进蛋白质合成;③促进脂肪降解;④减少外周组织对葡萄糖的利用;⑤促进水、矿物质代谢;⑥还有抗衰老,促进脑功能效应,增强心肌功能,提高免疫功能等作用。
(4)类胰岛素生长因子(IGF-1):IGF-1为肝脏对GH反应时产生的一种多肽,由70个氨基酸组成,基因定位于第12号染色体长臂,含有6个外显子。血中90%的IGF-1由肝脏合成,其余由成纤维细胞、胶原等细胞合成。IGF-1的生理作用主要为刺激软骨细胞增殖、分化和胶原的合成。肝脏合成的IGF-1在血中与类胰岛素生长因子结合蛋白(IGFBPs)结合,输送到外周组织发挥作用,软骨细胞、成纤维细胞、肌肉细胞、血管内皮细胞均存在IGF受体。
(5)下丘脑-GH-IGF轴:下丘脑-GH-IGF轴是调控人体生长的主要内分泌系统。GH合成和分泌受下丘脑生长激素释放激素(GHRH)和生长激素释放抑制激素(SS)双重控制。GHRH除促进分泌GH外,亦增加细胞内mRNA,促进GH合成。GHRH亦呈脉冲式分泌,其机制较复杂,可能主要受中枢神经系统的多种神经递质和神经肽的调节。动物实验证实垂体中存在GHRH受体。GHRH和SS共同调节GH的释放。SS由14个氨基酸组成,它抑制GHRH的产生,两者均与垂体前叶特异性受体结合,其分泌亦受中枢神经系统的多种神经递质和神经肽的调节。GH分泌受应激、
低血糖、运动的影响,促使其分泌增加,内分泌激素如雌激素、睾酮、甲状腺素亦促使其分泌。而高血糖、游离脂肪酸可抑制GH的分泌。
生长激素释放肽(GHRPs)是近年发现,并且能人工合成的促GH释放肽,它通过不同于GHRH的作用方式刺激垂体释放GH。GHRP-6是第一个被发现的GHRP,其衍生物为Hexarelin,静脉注射可促进GH分泌,已有部分研究报告发表。
2.生长障碍有关基因 生长是一种极为复杂的过程,需要基因的表达调控及细胞的分裂和增殖。人的生长和最终身高受遗传因素、先天因素、出生时体重和身高、营养和激素等因素共同作用。随着内分泌分子生物学的研究进展,近年发现了一些能导致矮小的基因,导致生长障碍伴基因突变的部分疾病见表1。
临床表现
临床表现:
生长激素缺乏患儿特别是没有先天性头颅畸形的儿童,于出生时身长和体重多正常,而有GH不敏感或GH受体缺陷的患儿出生长度可低于正常。严重GH缺乏时如GHD基因缺失,1岁时即可明显矮于正常平均值的-4SD 。
GH缺乏症的部分患儿出生时有难产史、窒息史或者胎位不正,以臀位、足位产多见。出生时身长正常,出生后5个月起出现生长减慢,1~2岁明显。多于2~3岁后才引起注意。随年龄的增长,生长缓慢程度也增加,体型较实际年龄幼稚,四肢和身体比例匀称。自幼食欲低下。典型者矮小,皮下脂肪相对较多,腹脂堆积,圆脸,前额略突出,小下颌,上下部量正常、肢体匀称,高音调声音。学龄期身高年增长率不足4cm,严重者仅2~3cm,身高偏离在正常均数-2SD以下。患儿智力正常。出牙、换牙及骨龄均延迟。
青春发育大多延缓。伴有垂体其他促激素不足者,多为缺乏促性腺激素(gonadotrophic hormone),表现为没有性发育,男孩
小阴茎、小睾丸,女孩乳房不发育,原发
闭经;若伴有ACTH缺乏,则常有皮肤色素沉着和严重的
低血糖表现;伴有促甲状腺激素不足,则表现为甲状腺功能低下。部分病例伴有多饮多尿,呈部分性
尿崩症。
鉴别诊断
1.宫内发育迟缓 通常将足月儿体重低于2.5kg者诊断为宫内发育迟缓(intrauterine growth retardation,IUGR)。目前本症可分为两类:一类是普通型的IUGR,表现为匀称性矮小,另一类是不对称身材矮小(Russell-Silver综合征)。内分泌功能检测一般正常,无
生长激素缺乏。
普通型的IUGR无性别差异,除匀称性矮小外不伴有畸形。表现有消瘦、纤弱,腹部脂肪堆积;食欲一般,三角脸,小下颌,前额宽大,性发育异常,骨龄往往延迟。
男女均可能患Russell-Silver征,除出生体重低和身材小外,常伴多种畸形或发育异常,如:
(1)单侧肢体肥大。
(3)颅面骨发育异常,小脸,三角脸,亦可有第五指短小弯曲,并趾等。
2.体质性生长和发育延迟 体质性生长和发育延迟或体质性矮小(constitutional delay of growth and puberty)多见于男孩,在儿童矮小症中占1/3以上。其父母可有青春期发育延迟的历史。性发育延迟愈明显者,家族史往往愈显著。患儿内分泌功能检测一般皆正常,但GH水平经药物激发后,可呈部分缺乏或暂时性缺乏现象。但延迟出现的
自发性青春期发育,仍可能使其终身高和性成熟达到正常水平,故此类患儿属正常生长发育中的一种变异。
3.特发性矮小 特发性矮小(idiopathic short stature)需排除所有已知的病因,无器质性疾病,患儿出生时身高、体重正常,矮小匀称。其GH自然分泌(生理性分泌)及药物激发后的峰值在正常范围内。通常矮小并不严重,可在-2.2(±0.6)SD水平,身高增长的速度可近似正常儿童或稍偏缓,其他内分泌激素及生化指标均无明显改变,亦无青春发育延迟。近年来有人试用GH治疗,认为近期身高增长虽略加速,但终身高仍不能达到标准。故对正常无GH缺乏的家族性矮小,有无必要使用长程昂贵的GH药物治疗尚有争议。
4.营养缺乏性矮小 营养缺乏性生长迟缓(nutritional growth retardation)或营养性矮小最主要的病因是因贫穷而营养摄入不足,但亦见于因主观自限饮食,摄取营养不合理导致生长受累。患儿体重虽较同龄儿低,但其体重/身高之比常与非营养性矮小(家族性矮小、体质性矮小)者相似,故难以区别。器质性疾患或非器质性疾病均可导致营养缺乏性生长迟缓。营养缺乏生长迟缓属暂时性,恢复足够营养摄入并调整饮食结构使之合理,则生长可加速。
5.精神、心理障碍性矮小 精神、心理障碍性矮小(psychosocial short stature)常发生在有父母感情不和,离异家庭或单亲子女家庭,患儿精神心理受挫,影响了下丘脑-GH-IGF轴功能,GH分泌可正常或缺乏。本症机制复杂,可能与慢性营养缺乏及GH神经分泌功能紊乱有关。
治疗
治疗:目前对
生长激素缺乏症的治疗主要采用GH替代治疗。无论特发性或继发性GH缺乏性矮小均可用GH治疗。开始治疗年龄越小,效果越好。但是对颅内肿瘤术后导致的
生长激素缺乏症患者或者白血病患者需慎用。
1.治疗剂量 目前多数学者推荐每周使用剂量为0.5~0.7U/kg,每晚临睡前皮下注射0.1U/kg。最大效应是在开始初6~12个月。上海市儿科医学研究所采用进口GH治疗20例,其生长速率从原<4cm/年加速到9.2~13.7cm/年,平均每年增长12cm。国产GH治疗效果类同进口药物。
2.应用方法 采用皮下注射,药物达到峰值时间为2~4h,血液清除时间为20~40h。可选择在上臂、大腿前侧和腹壁、脐周等部位注射。
3.GH治疗并发症
(1)局部反应。
(2)抗体产生。
(3)低甲状腺素血症。
(4)血转氨酶升高,一般表现轻度升高,随药物停用而逐渐消失。
随着
重组人生长激素(rhGH)的大量供应,rhGH的治疗病种已经超越了
生长激素缺乏症,Turner综合征采用
生长激素治疗显示出一定的疗效。每周rhGH使用剂量稍大,为1.0U/kg,治疗从小年龄开始效果较好,若患者年龄大于14岁,年生长速率小于2.5cm应停止治疗。
蛋白同化类固醇药物可促进生长,但是该类药物可明显加速骨龄发育,加快骨骺融合,对最终身高无明显改善。